
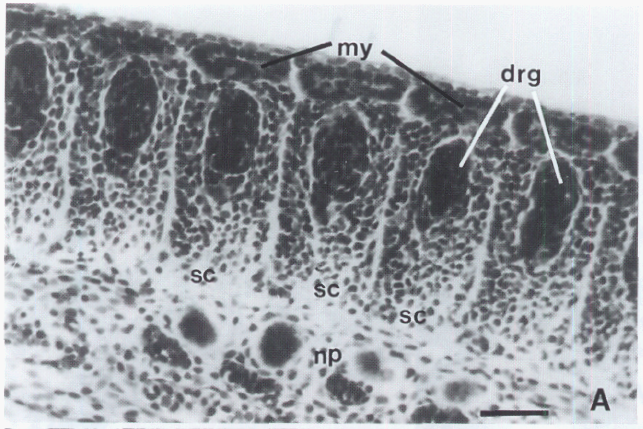
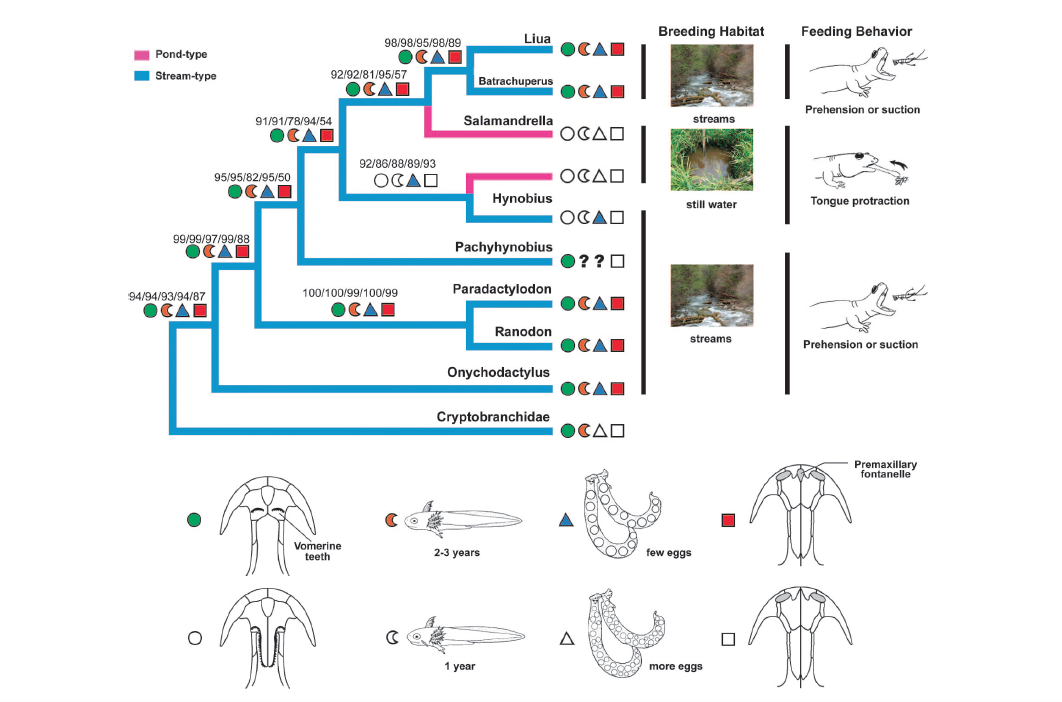
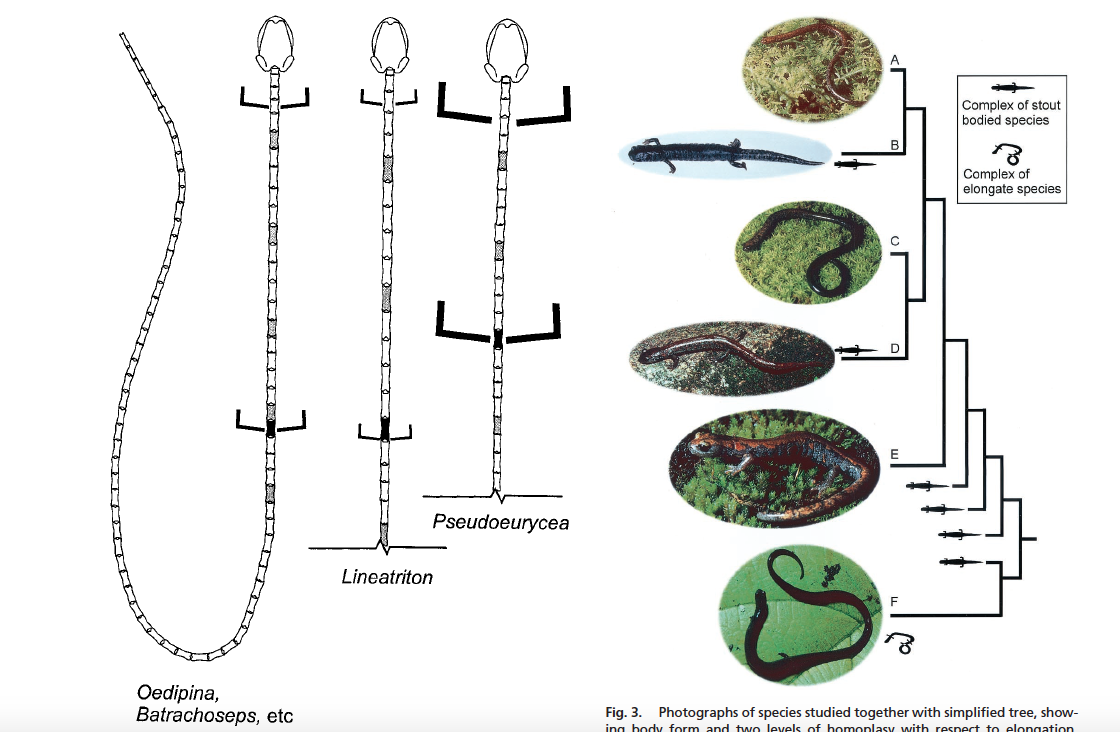

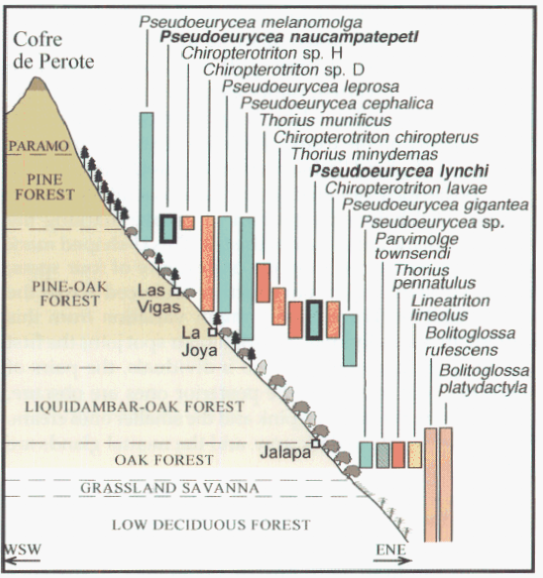
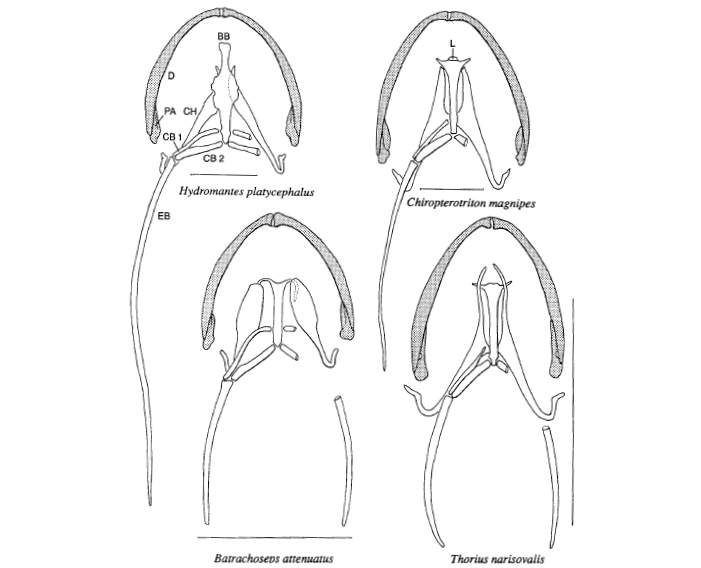
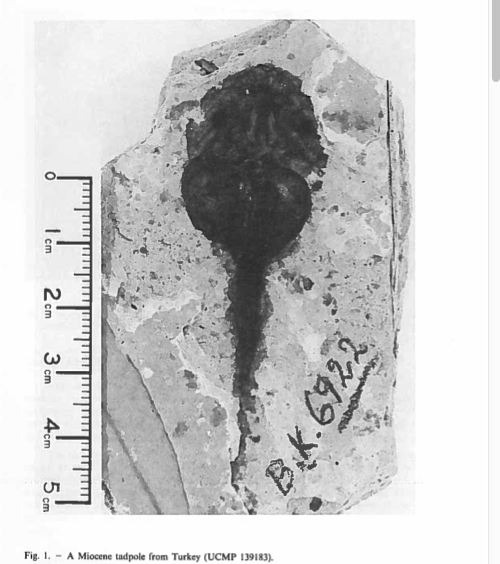
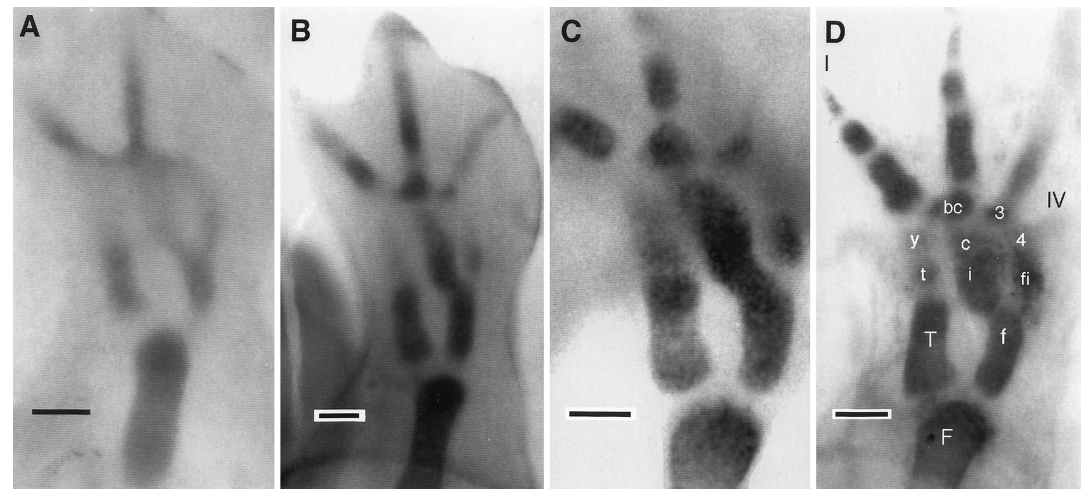
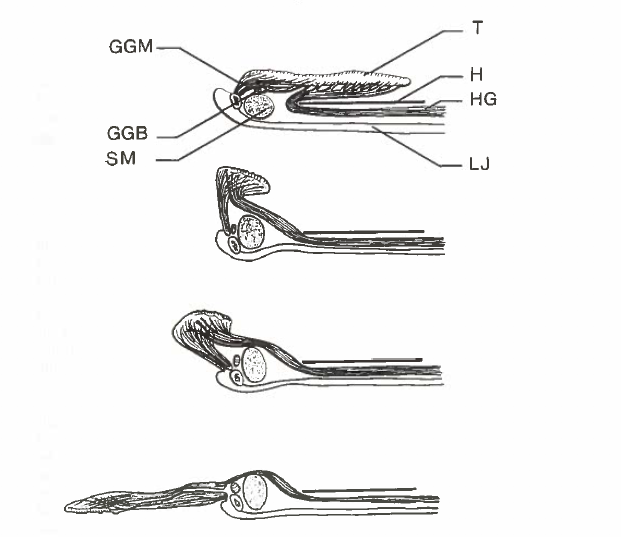
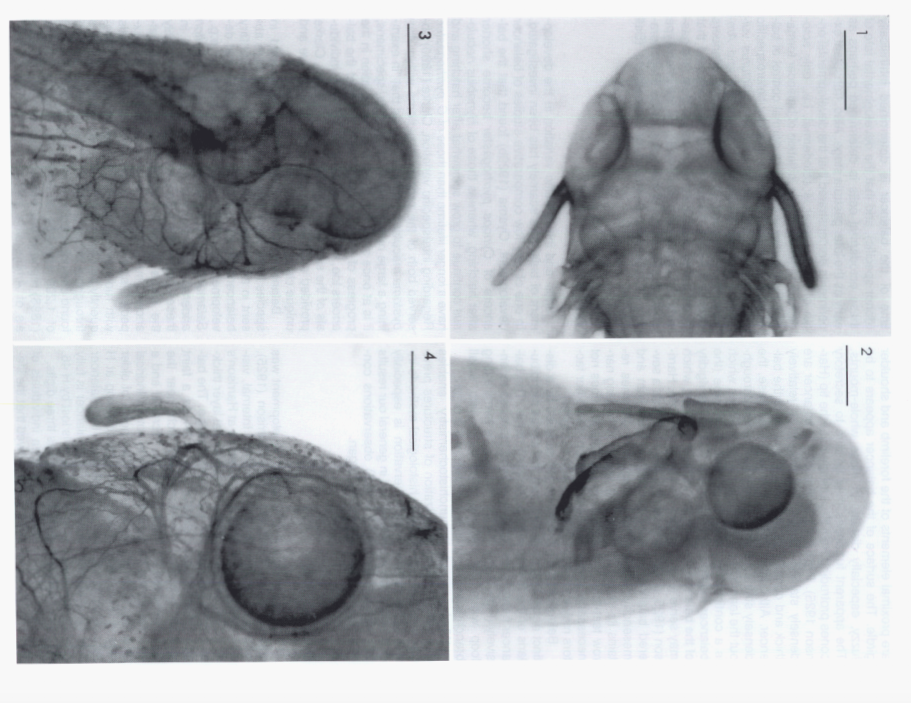
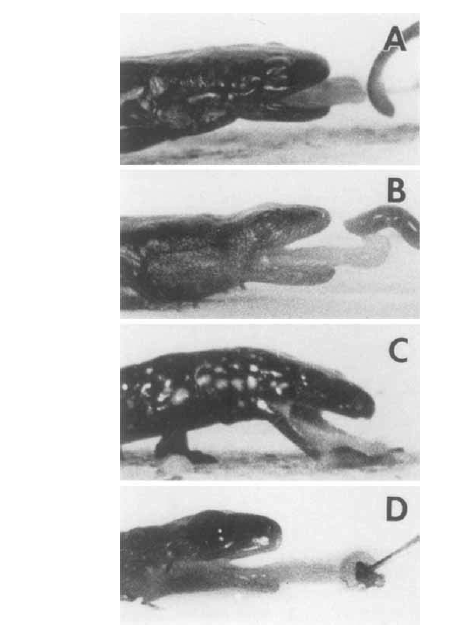

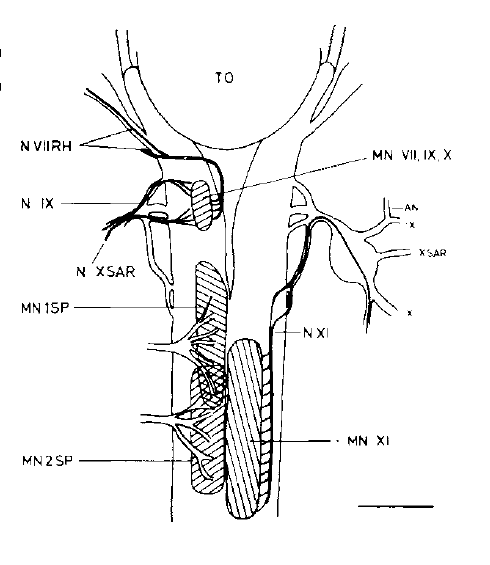

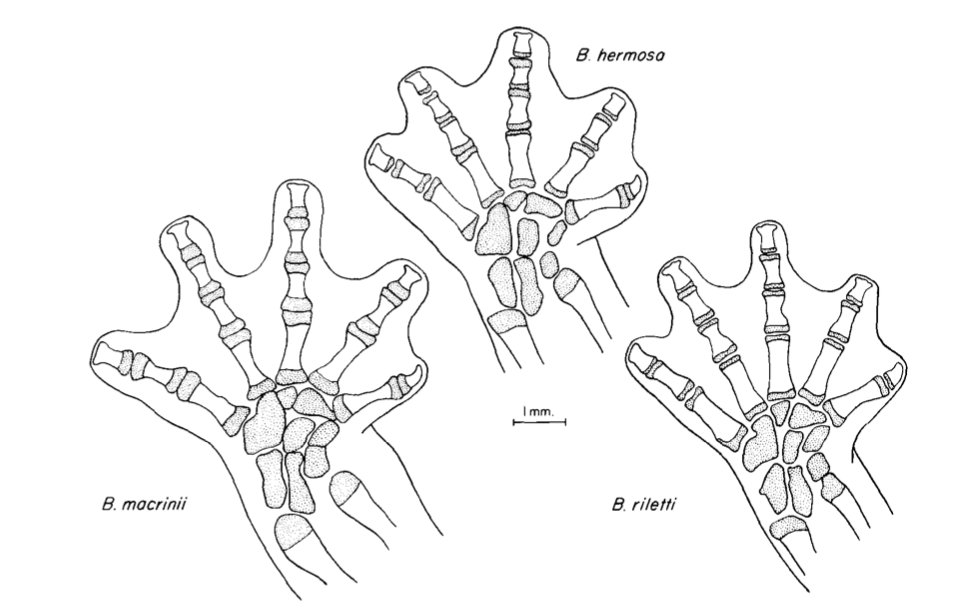
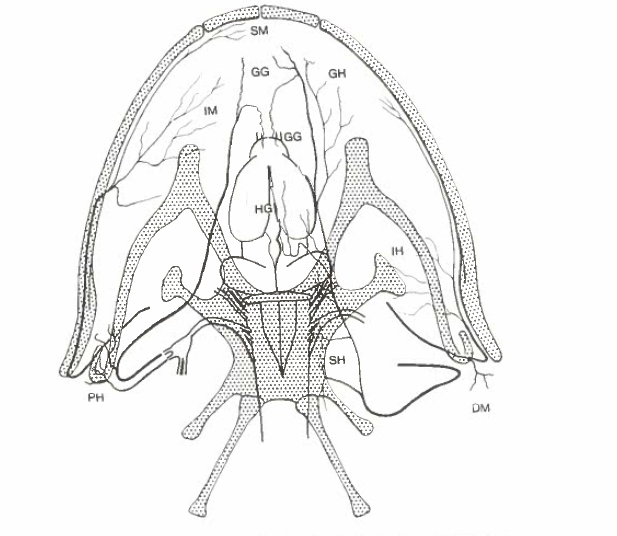
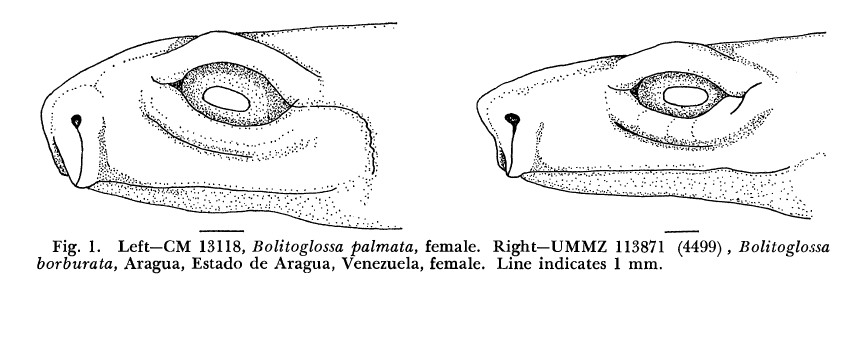
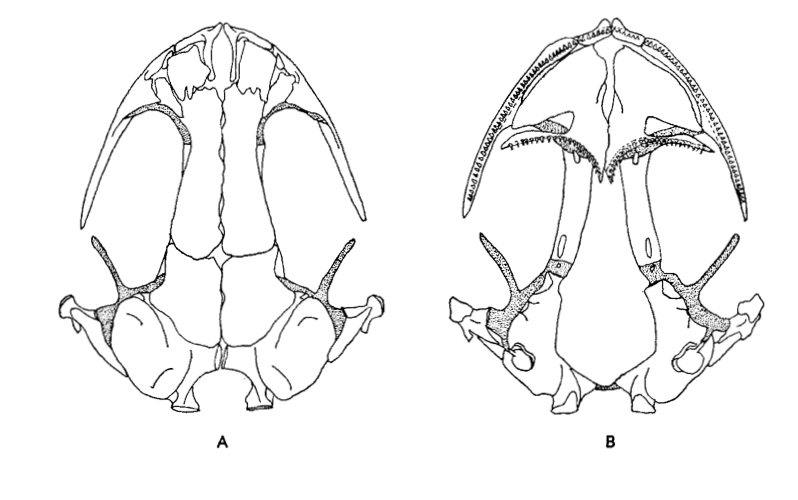
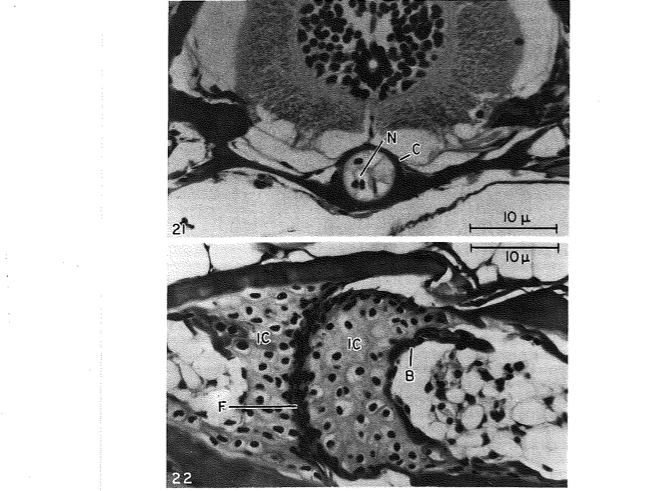

David Wake passed away the morning of April 29, 2021, surrounded by his family. A native of South Dakota, he earned his Ph.D. at the University of Southern California where he met his wife, Marvalee. He began his career at the University of Chicago, and then joined the faculty of UC Berkeley in 1968. He was the Director of the Museum of Vertebrate Zoology for 27 years (1971-1998). He served as President of the Society for the Study of Evolution, the American Society of Naturalists, and the American Society of Zoologists. Dave, as he was known, was a towering figure in evolutionary biology and herpetology, and trained generations of students, including many leaders in the field today. Dave was a member of the US National Academy of Sciences and was recognized by many awards. His deep wisdom, gentle demeanor, and friendship were an inspiration to all. We miss him deeply.